

Background: Intensive aquaculture in southern Spain is growing, leading to increased use of plant-based in- gredients in fish feed. While this shift results in more nutritionally balanced feeds, it also introduces the risk of contaminants like mycotoxins and pesticides, which can negatively affect feed production, fish health, and pose potential risks to consumers. However, studies on these contaminants in aquaculture feed are limited. Results: In this study, a multiclass method was developed to determine 27 mycotoxins and 11 pesticides in aquaculture feed by ultrahigh performance liquid chromatography coupled with high resolution mass spec- trometry (UHPLC-HRMS). The method involved a previous ultrasound-assisted solid-liquid extraction using an acidified mixture of acetonitrile and water, followed by a dispersive solid-phase extraction with MgSO4 and C18. Characterization of the method was satisfactory for almost all compounds, with recovery values between 70 and 120 % and relative standard deviations (RSDs) below 20 %. The quantification limits were comparable to those reported in studies focusing on a single family of compounds. The method was applied to the analysis of aquaculture feeds with diverse compositions, revealing a high incidence of the mycotoxins enniatins, as well as the pesticides tebuconazole and pirimiphos methyl. Conclusion: The possible source of these contaminants and residues was investigated by analysing the main raw materials of the feeds, attempting to link their origin with the wheat and soy. Additionally, non-targeted analysis identified six additional pesticides, and one pesticide adjuvant not included in the targeted analysis, highlighting the importance of such studies.


Categories
- Amino acids (2)
- Animal feed (7)
- PTSO (3)
- Anthelmintic (6)
- Benzimidazoles (6)
- Antibiotics (31)
- 5-nitroimidazoles (10)
- Aminoglycosides (2)
- Quinolones (8)
- Sulfonamides (2)
- Tetracyclines (6)
- β-lactams (5)
- Cephalosporins (3)
- Biological fluids (13)
- Human serum (2)
- Pig serum (3)
- Urine (8)
- Biomonitoring (1)
- C4D (1)
- Chemiluminescence (6)
- Photoinduced (2)
- Chromatography (62)
- Capillary HPLC (12)
- GC (1)
- HILIC (5)
- HPLC (5)
- UHPLC (36)
- Contaminants (4)
- Cyclodextrins (1)
- DART (1)
- Drugs (8)
- Anaesthetics (1)
- Muscle relaxants (1)
- Steroids (1)
- Electrochromatography (1)
- Electrophoresis (41)
- Exposomics (2)
- Fingerprints (1)
- Flow injection analysis (1)
- Fluorescence (15)
- LIF (7)
- Photo-induced (3)
- Food (66)
- Aquaculture products (4)
- Barley (4)
- Cheese (1)
- Eggs (5)
- Fruit (2)
- Dried fruit (1)
- Honey (4)
- Infant foods (2)
- Juice (3)
- Maize (3)
- Meat (4)
- Chicken muscle (1)
- Pig kidney (1)
- Pork muscle (1)
- Milk (12)
- Nuts (2)
- Oat (2)
- Pseudocereals (1)
- Rice (6)
- Seeds (2)
- Spelt (1)
- Supplements (5)
- Blue-green algae (1)
- Pollen (2)
- Propolis (1)
- Royal jelly (1)
- Vegetable milks (2)
- Vegetables (6)
- Aromatic herbs (1)
- Celery (1)
- Cucumber (1)
- Lettuce (1)
- Milk thistle (1)
- Pepper (1)
- Spinach (1)
- Tomato (1)
- Wheat (7)
- Durum wheat (1)
- Wine (4)
- Yogurt (2)
- Human biomonitoring (1)
- Insects (1)
- Honeybees (1)
- Ionic liquids (1)
- Mass spectrometry (54)
- Ion mobility (4)
- Metabolites (1)
- Destruxins (1)
- Metabolomics (4)
- Pesticides (22)
- Carbamates (9)
- Fipronil (3)
- Neonicotinoids (4)
- Polar (1)
- Sulfonylurea (3)
- Pharmaceutical formulations (1)
- Plants (1)
- Potato plant (1)
- Projects (66)
- AGL2015-70708-R (21)
- B-AGR-202-UGR20 (4)
- EQC2018-004453-P (2)
- P12-AGR-1647 (26)
- PID2020–120020RA-I00 (4)
- PID2021-127804OB-I00 (8)
- PROYEXCEL_00195 (10)
- RED2018-102522-T (2)
- RED2022-134079-T (1)
- RTI2018-097043-B-I00 (9)
- RYC2023-044255-I (1)
- Review (11)
- Sample treatment (90)
- DLLME (16)
- Dilute and shoot (2)
- Hollow-fiber liquid-phase microextraction (1)
- Liquid-liquid extraction (1)
- MIPs (8)
- NADES (2)
- Protein precipitation (1)
- QuEChERS (24)
- QuPPe (1)
- SALLE (16)
- SPE (16)
- Solid-liquid extraction (4)
- UASEME (1)
- USLE (1)
- automated SPE (1)
- dSPE (1)
- on-line SPE (1)
- Sequential injection analysis (1)
- Soil (1)
- Theses (5)
- Toxins (34)
- Cyanotoxins (6)
- Mycotoxins (28)
- Aflatoxins (4)
- Enniatins (2)
- Ergot alkaloids (4)
- Trabajo fin máster (2)
- UV-vis (29)
- Water (22)
- Thermal spring (1)
- Wastewater (1)
Archives
- November 2025
- September 2025
- June 2025
- February 2025
- January 2025
- December 2024
- October 2024
- July 2024
- April 2024
- January 2024
- December 2023
- October 2023
- September 2023
- August 2023
- July 2023
- February 2023
- September 2022
- April 2022
- November 2021
- September 2021
- July 2021
- June 2021
- April 2021
- December 2020
- October 2020
- August 2020
- April 2020
- March 2020
- February 2020
- December 2019
- July 2019
- June 2019
- February 2019
- November 2018
- October 2018
- September 2018
- June 2018
- May 2018
- March 2018
- February 2018
- November 2017
- October 2017
- August 2017
- July 2017
- June 2017
- May 2017
- April 2017
- March 2017
- February 2017
- January 2017
- December 2016
- November 2016
- September 2016
- August 2016
- July 2016
- May 2016
- March 2016
- February 2016
- December 2015
- November 2015
- August 2015
- October 2013
- September 2013
- August 2013
- July 2013
- May 2013
- February 2013
- January 2013
- December 2012
- November 2012
- August 2012
- July 2012
- June 2012
- February 2012
- December 2011
- November 2011
- September 2011
- July 2011
- April 2011
- September 2010
- July 2010
- June 2010
- May 2010
- April 2010
- February 2010
- January 2010
-
Recent Posts
- Simultaneous determination of mycotoxins and pesticides by UHPLC-HRMS in aquaculture feed from intensive farms
- A greener approach for determining cyanotoxins in vegetables: From NADES-air-assisted dispersive liquid-liquid microextraction to capillary electrophoresis–mass spectrometry analysis
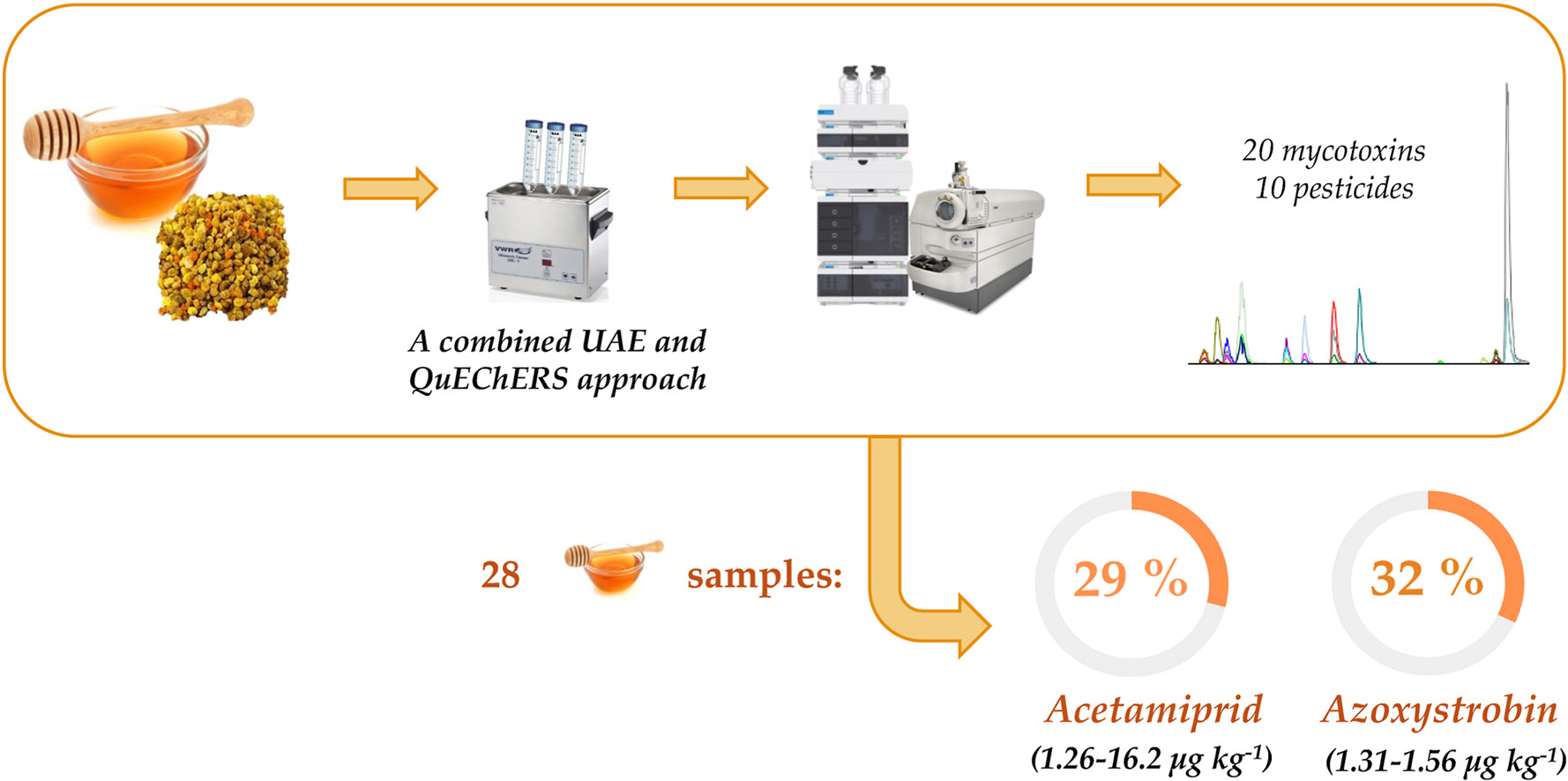
- Addressing chemical risks in beekeeping products: development of a combined ultrasound-assisted extraction and QuEChERS-based method for the simultaneous determination of mycotoxins and pesticides in bee pollen and honey.
- Toxic cyanopeptides monitoring in thermal spring water by capillary electrophoresis tandem mass spectrometry.
- Liquid chromatography-tandem mass spectrometry for the determination of multiple mycotoxins in serum through suspect screening and targeted approaches: Advancing human mycotoxin biomonitoring.
Tags
